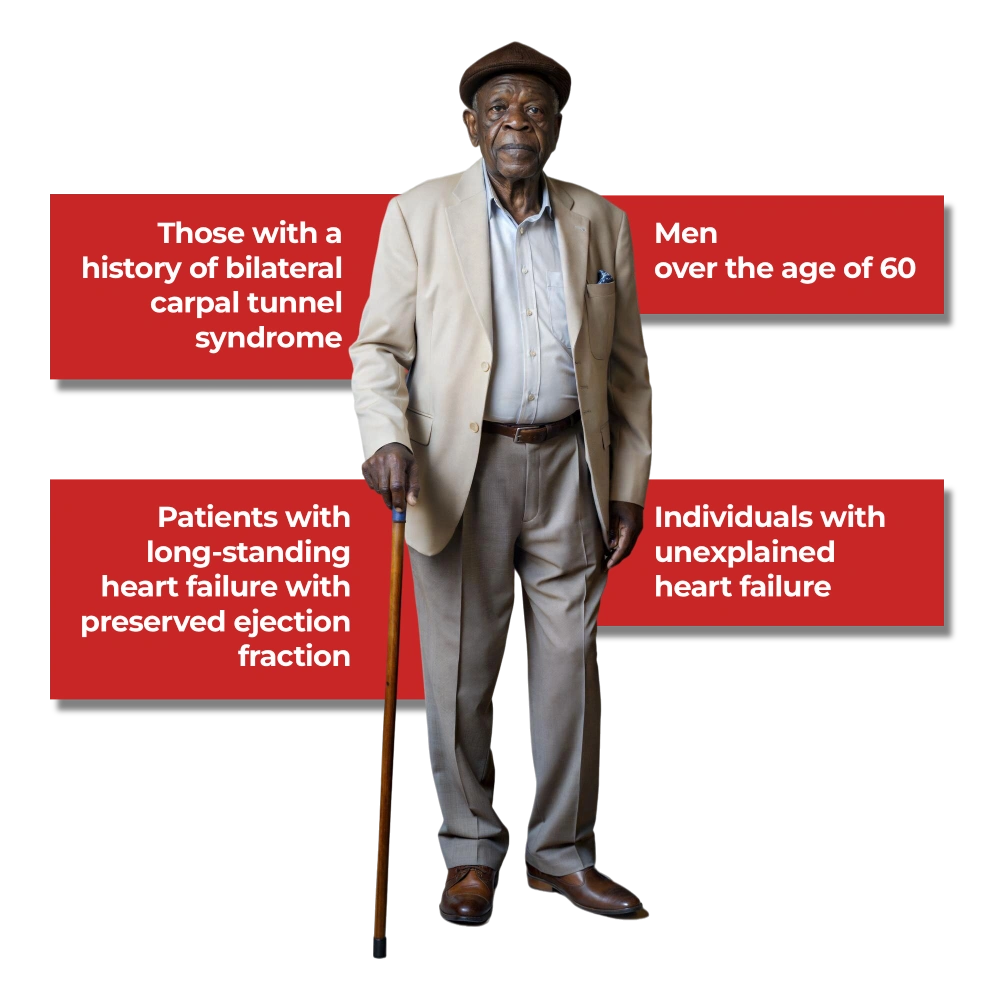
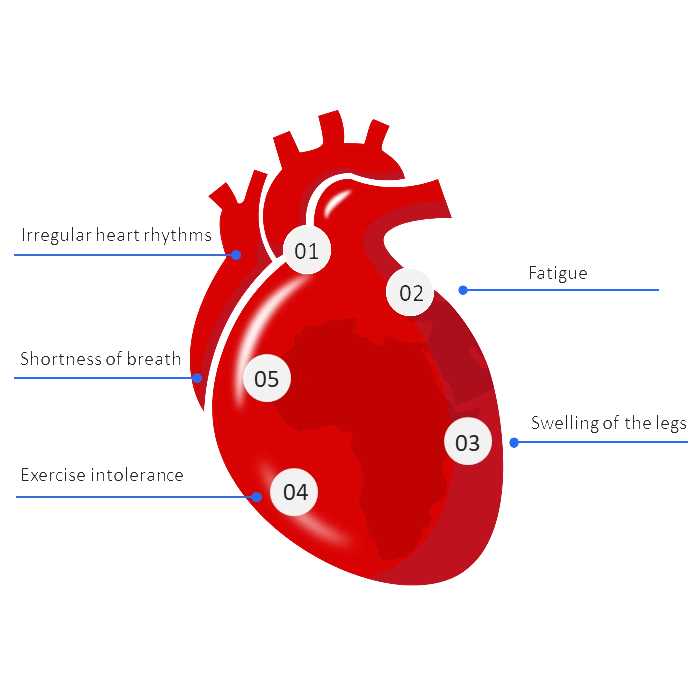
Wild-type ATTR amyloidosis is likely underdiagnosed across Africa. Aging populations, increasing rates of cardiovascular disease, and limited awareness create a setting where many patients may be misclassified as having routine heart failure.
Improving awareness among cardiologists, internists, and general practitioners is essential to identifying patients earlier and improving outcomes.
Amyloidosis Africa works to increase recognition of wild-type ATTR amyloidosis through medical education, research collaboration, quality improvement initiatives, and advocacy across the continent.