Hereditary ATTR amyloidosis is caused by mutations in the transthyretin (TTR) gene, and certain variants have been identified more frequently in people of African ancestry.
One of the most well-known variants is Val122Ile (V122I), also referred to as p.Val142Ile in updated genetic nomenclature. This mutation is particularly common among individuals of West African descent and in African diaspora populations. Studies published in the New England Journal of Medicine have shown that approximately 3 to 4 percent of African Americans carry this variant, making it one of the most prevalent amyloid-associated mutations worldwide.
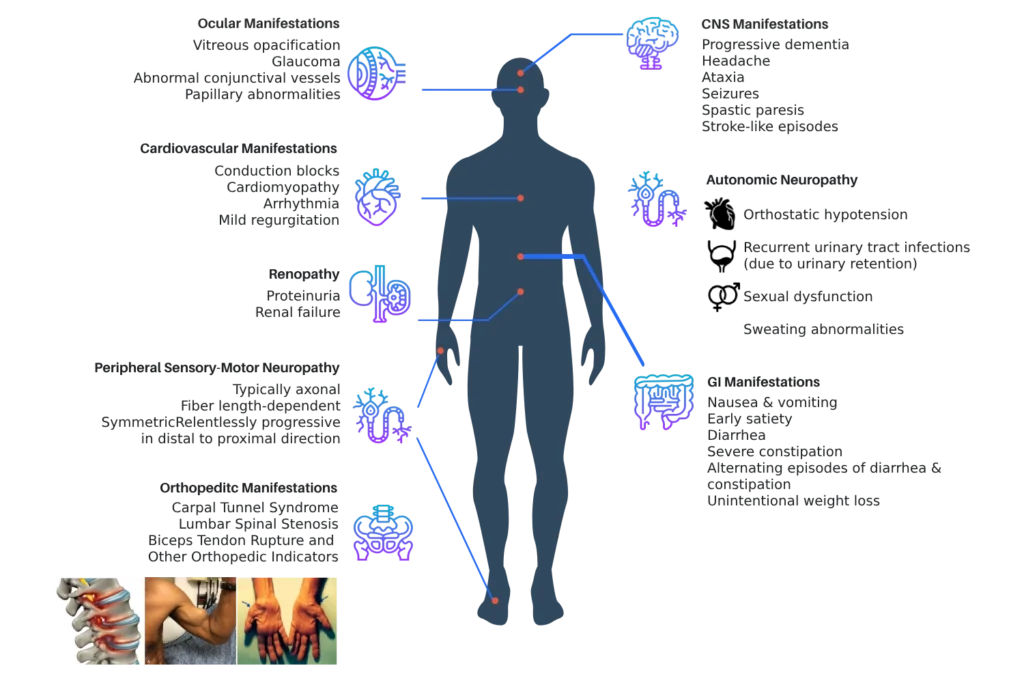
The Val122Ile mutation primarily affects the heart and is strongly associated with ATTR cardiac amyloidosis. It often presents later in life and may be misdiagnosed as hypertensive heart disease or heart failure with preserved ejection fraction. Because symptoms overlap with common cardiovascular conditions, many affected individuals remain undiagnosed.
While Val122Ile is the most recognized ATTR mutation associated with African ancestry, it is not the only one. Genetic diversity across African populations is extensive, and additional TTR variants may exist that are underreported or insufficiently studied due to limited access to genetic testing and research infrastructure in many African countries.
A comprehensive scientific overview of hereditary ATTR amyloidosis, including genetic variants and clinical manifestations, is available in GeneReviews.
Understanding ATTR mutations in African populations is essential because it:
Improves clinician awareness when evaluating unexplained cardiomyopathy
Supports targeted genetic screening strategies
Enables family counseling and early monitoring
Strengthens African representation in global amyloidosis research
Importantly, carrying a TTR mutation does not automatically mean a person will develop severe disease. Age of onset, clinical presentation, and progression can vary significantly, even among individuals with the same mutation.
Expanding access to genetic services, structured screening programs, and research collaboration across Africa is critical to identifying affected families earlier and improving outcomes on the continent.