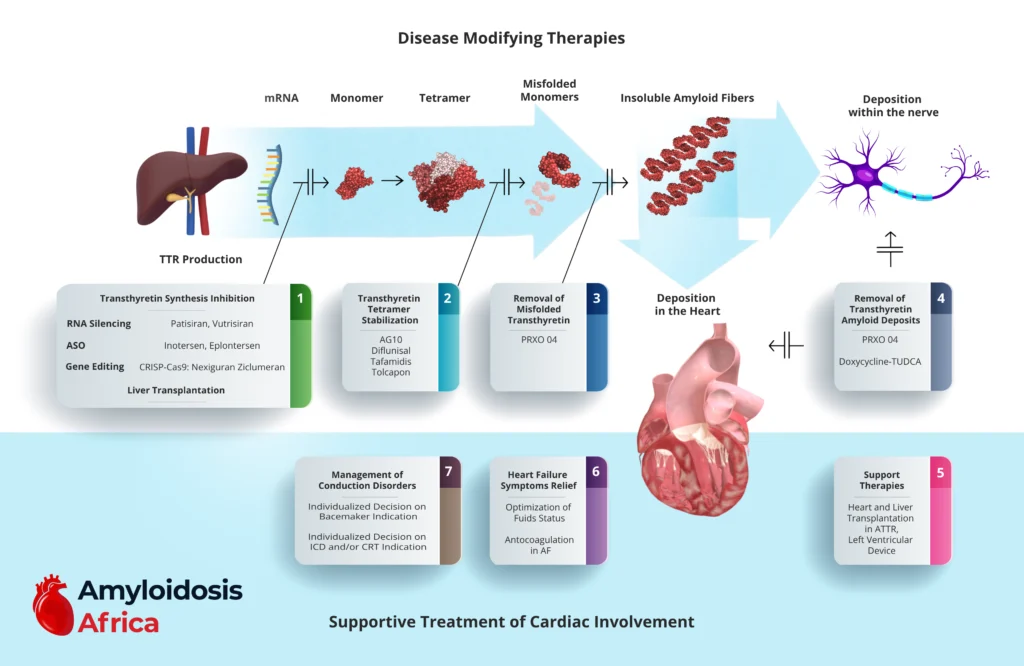
1. Inhibition of TTR Production (Gene Silencing & Editing)
These therapies reduce hepatic production of transthyretin, lowering circulating TTR levels and slowing amyloid formation.
RNA Interference (RNAi)
These small interfering RNA therapies target TTR mRNA in hepatocytes, significantly reducing serum TTR levels.
Antisense Oligonucleotides (ASO)
These inhibit TTR mRNA translation, reducing protein synthesis.
Gene Editing
Emerging therapies aim to permanently silence the TTR gene through in vivo genome editing.
Liver Transplantation
Historically used in hereditary ATTR (hATTR), liver transplantation removes the primary source of mutant TTR production. However, it is now less common due to targeted pharmacologic therapies.
2. Transthyretin Tetramer Stabilization
Tetramer dissociation is the rate-limiting step in amyloid formation. Stabilizers bind TTR and prevent dissociation into misfolded monomers.
Approved and Investigational Stabilizers
Tafamidis has demonstrated mortality and hospitalization benefits in ATTR-CM and is currently the cornerstone disease-modifying therapy.
3. Removal of Misfolded Transthyretin
Emerging therapies aim to enhance the clearance of circulating misfolded TTR species.
These therapies seek to neutralize toxic TTR aggregates before tissue deposition.
4. Removal of Amyloid Deposits
Therapies targeting established amyloid fibrils aim to reverse organ dysfunction.
Amyloid removal remains an area of active research and represents a promising future direction.
Supportive Treatment of Cardiac Involvement
Even with disease-modifying therapy, most patients require careful cardiac management.
5. Heart Failure Management
Heart failure in ATTR-CM is predominantly restrictive with preserved or mildly reduced ejection fraction.
Key Principles:
Careful fluid management
Loop diuretics as first-line therapy
Mineralocorticoid receptor antagonists when tolerated
Avoid excessive preload reduction
Caution:
ACE inhibitors, ARBs, and beta-blockers are often poorly tolerated due to low stroke volume.
Calcium channel blockers and digoxin should generally be avoided due to potential toxicity in amyloid hearts.
Anticoagulation
Atrial fibrillation is common and carries a high thromboembolic risk. Anticoagulation is often recommended regardless of CHA₂DS₂-VASc score.
6. Conduction System Disease Management
ATTR frequently affects the conduction system.
Management includes:
Pacemaker implantation for symptomatic bradycardia or AV block
Individualized decision-making for ICD placement
Consideration of CRT in selected patients
Electrophysiologic monitoring is essential due to progressive conduction abnormalities.
7. Advanced Supportive Therapies
Heart Transplantation
May be considered in carefully selected patients, particularly younger individuals with hereditary disease.
Combined Heart–Liver Transplantation
Rare but considered in selected hereditary cases.
Mechanical Circulatory Support
Left ventricular assist devices (LVAD) have limited use due to restrictive physiology but may be considered in specific contexts.